
In a remarkable discovery, researchers have found that a rare and relatively unknown disease shares a fundamental mechanism with the well-known condition, cystic fibrosis. This unexpected connection sheds light on the intricate pathways that govern both diseases and could have significant implications for treatment strategies.
Researchers from the University of Michigan have discovered that the same thing happening in a type of cystic fibrosis also occurs in a rare disease called cystinosis.
There’s a process in our cells that helps clean up messed-up proteins. But this process goes wrong in cystinosis, a rare illness caused by genes. This causes something called cystine crystals to gather in the cell. These crystals make the cells not work well, affecting our body parts like kidneys and eyes.
The issue starts with a part of the cell called the lysosome. It’s like the cell’s recycling center. It takes in things the cell doesn’t need anymore, breaks them into functional parts, and uses them again.
When a protein that helps carry back a reused building block for the cell gets messed up because of a change in its structure, the lysosome can’t do its job. This makes a mechanism in the cell to clean up that bad protein. But because of this cleanup, a cystine building block starts to pile up in the lysosome, causing trouble.
Varsha Venkatarangan, a graduate student in the U-M Department of Molecular, Cellular, and Developmental Biology and lead author of the study, said, “If cystinosis is not treated at an early age, some of the effects are irreversible, and it could include impaired growth, kidney failure, and neurological problems. Typically, the symptoms of the disease are treated rather than the root problem. So we have been wondering what could be the possible cellular mechanism of this disease.”
Venkatarangan worked with special cells called fibroblasts, taken from the skin of people with a particular illness. She discovered that a cell process called endoplasmic-reticulum-associated degradation, or ERAD, breaks down a messed-up protein version that helps transport things in the cell.
This ERAD process is the same one that causes other illnesses, like a significant kind of cystic fibrosis. And since they knew about this process, the researchers could use specific drug molecules to stop the broken protein from getting destroyed. They shared their discoveries in the Journal of Clinical Investigation.
One of these drug molecules is like a helper for the protein. It helps the messed-up version of the protein fold correctly and stay strong instead of breaking down.
The researchers, led by Professor Ming Li, got interested in this protein that transports things and wanted to know why changes in its genes caused the illness. They looked at nearly 40 changes that caused the disease and saw that one change made the protein break down quickly, unlike the normal version.
“We wanted to know why this disease-causing change in the protein was breaking down so quickly and how it was happening,” Venkatarangan explained.
To find out, Venkatarangan did many tests using genes and chemicals to stop different parts of the cell from breaking down proteins. She figured out that a process called ERAD was the reason. ERAD is like a pathway inside a part of the cell called the endoplasmic reticulum. This part looks like a network of tiny tubes and sheets that help move the cell’s proteins where they need to go. ERAD’s job is to find improperly folded proteins and mark them for breaking down.
In a person without illness, the protein helps move called cystine out of a part of the cell called the lysosome (shown on the left in the picture). But in someone with an illness, the broken protein gets broken down by a process called ERAD, which leads to a cystine buildup in the lysosome (shown in the middle of the picture). However, using a chemical used for treating another illness called cystic fibrosis, the researcher protected the broken protein from getting destroyed (shown on the right in the picture). This chemical helped the lysosome get rid of the extra cystine.
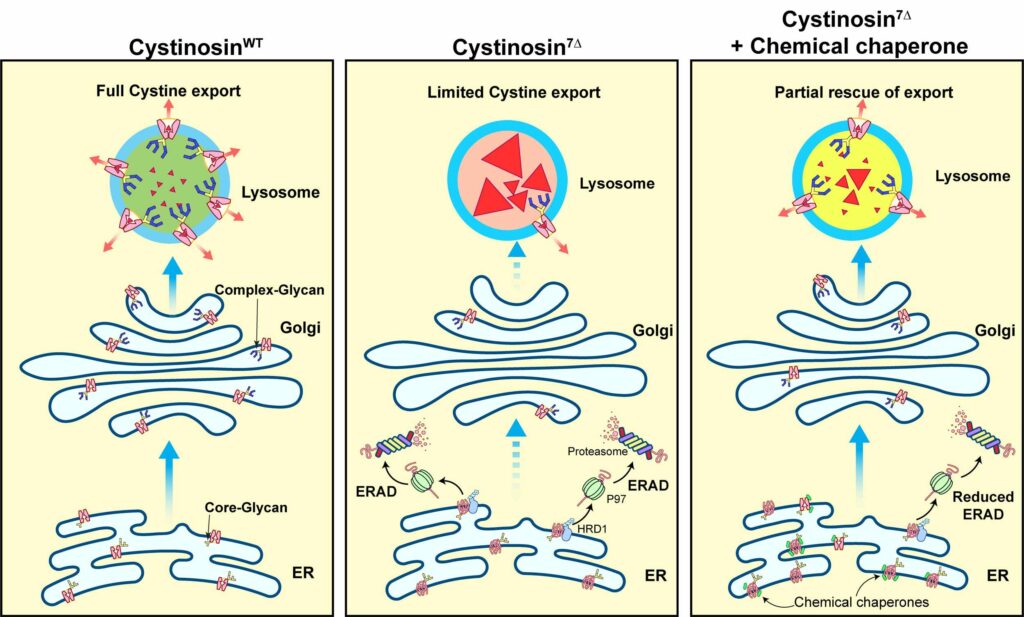
Since this process was already known in other illnesses like cystic fibrosis, researchers had already found drug molecules that could help with ERAD. These drugs are called chemical chaperones, and they allow the protein to fold correctly so that it works better.
“Imagine this small chemical chaperone like a magic helper. It somehow sticks to our messed-up protein and stops it from breaking down,” said Li.
The researchers made the protein more stable and checked if it was working in the right place to move things out of the lysosome. To test this, they worked with another group of researchers and measured how much cystine was in the lysosome. If the protein was working, there should be less cystine.
“The buildup went down a lot – it reduced by almost 70%,” Li explained. “This almost brought things back to normal levels in the system. Suppose we use this special helper chemical to treat patients. In that case, we might be able to reduce the extra cystine in the lysosome significantly.”
The endoplasmic reticulum impacts how lysosome membrane proteins function, as these proteins are transported from the endoplasmic reticulum. During the creation of the lysosome, it requires particular proteins to facilitate its functioning. These proteins travel through the endoplasmic reticulum and pass through the Golgi apparatus.
This Golgi apparatus packages the proteins and sends them to the lysosome. While they’re being moved around, these proteins have to fold correctly. If they don’t, they’ll be broken down early by systems that check if proteins are okay, like ERAD. The researchers found that these broken proteins disappear early during the moving process. They get stuck and broken down in the endoplasmic reticulum.
By repurposing a drug already being used, the team also achieved a goal set by the National Institutes of Health. This goal encourages finding new uses for drugs that are already approved, which can speed up the development of treatments. However, the team wants to clarify that their work is still in the early stages. They only tested it on cells from patients grown in the lab and have yet to try it on animals or humans.
“Remember, this is just a start, and we only tried it on cells from patients in a lab. So we can’t say how well it would work in real patients,” Venkatarangan said. “This also shows how important it is to treat each patient as an individual because often, we treat all people with the same illness in the same way. However, some mutations might need specific treatments. Understanding each patient’s unique mechanism could help us plan better ways to treat them.”
Journal Reference:
- Varsha Venkatarangan, Weichao Zhang, et al., ER-associated degradation in cystinosis pathogenesis and the prospects of precision medicine. The Journal of Clinical Investigation. DOI: 10.1172/JCI169551.